01执行摘要
总体结论:当前锁定工具栈大体稳健、方向正确,但有 5 处需要调整或重新评估,并有 6 个分析阶段仍是工具空白需补充选型。最关键的发现是——存在一篇同病种(晚发型 PE 胎盘)空间多组学论文,其空间代谢↔空间转录的配准与 MSI 处理方案,与本项目当前设定(elastix 同切片配准)不同,提供了直接可借鉴的实证范式。
✅ 经检验应保留 / 加强
- RCTD 空间去卷积——两个独立同行评审基准均列入最优梯队,且最快、支持 CPU 并行(无需 GPU)。建议同时跑 cell2location 交叉比对。
- Scanpy / squidpy / Snakemake / WGCNA / AUCell + GSVA / CMap 原理——主流程合理,保留。
🔼 应升级 / 调整
- 批次整合:scIB 基准显示 Harmony 在"复杂真实数据"非顶档;EVT/SCT 高异质场景考虑 scVI/scANVI(scvi-tools v1.4.3 活跃)或 Scanorama(稀有亚群)。需按 11 例样本规模实测
- 双细胞剔除:流程应新增该步骤,工具倾向 scDblFinder(而非已过时的 DoubletFinder)。
🔁 应重新评估 / 替换
- 空间配准:同病种 PE 论文用「相邻切片 + 自研 landmark spot-match(≥6 点缩放+旋转)」,而非 elastix 同切片仿射+B-spline。当前设定面临同场景反例,需评估。
- 空间域识别:以 BANKSY(统一细胞分型+组织域分割,多基准胜 SpaGCN)或 GraphST 增强/替换 SpaGCN。
➕ 填补空白(已有同病种实证)
- MALDI-MSI 处理/注释:Cardinal 3(R)+ pySM(METASPACE 引擎)——同病种 PE 空间代谢组研究已采用。
- 细胞注释参考图谱:Vento-Tormo 2018(~7 万细胞母胎界面图谱,含 EVT/SCT,亦是 CellPhoneDB 来源)。
- 滋养层分化轨迹:CellRank(速率核+连通核高维转移矩阵,更鲁棒)。
02分析阶段 → 推荐工具总览
17 个分析阶段的推荐主用工具与备选,及其对当前锁定工具栈的结论。状态列标明该行结论是否经本轮对抗式验证。
| # | 分析阶段 | 推荐主用 | 备选 | 对锁定栈 | 状态 |
|---|---|---|---|---|---|
| 1 | 单细胞 QC / 双细胞剔除 | scDblFinder R | DoubletDetection, Scrublet | 新增步骤 | ✓ |
| 2 | 批次整合 / 去批次 | scVI / scANVI Py (scvi-tools) | Scanorama;Harmony(小数据/简单批次) | 升级·条件 | ✓ |
| 3 | 细胞类型注释(滋养层亚群) | CellTypist + Vento-Tormo 2018 参考 Py | SingleR, Azimuth | 新增·参考图谱 | ✓图谱 |
| 4 | 滋养层分化轨迹 / RNA 速率 | CellRank 2 Py | scVelo, Monocle3, Palantir, PAGA | 新增 | ✓ |
| 5 | 细胞间通讯(EVT/SCT↔内皮/免疫) | CellPhoneDB / LIANA Py | CellChat, NicheNet | 候选 | ⚠ |
| 6 | Visium 空间去卷积 | RCTD R ✚ cell2location 交叉 Py | Tangram, CARD | 保留+交叉 | ✓ |
| 7 | 空间域识别 / 空间可变基因 | BANKSY RPy | GraphST, STAGATE;SpatialDE | 替换/增强 SpaGCN | ✓ |
| 8 | 空间转录↔空间代谢 配准 | landmark spot-match(相邻切片·同病种实证) | elastix / SimpleITK / ANTsPy(同切片) | 重新评估 | ✓ |
| 9 | MALDI-MSI 处理 / 代谢物注释 | Cardinal 3 R + pySM/METASPACE Py | SCiLS(商业), pyimzML | 填补空白 | ✓med |
| 10 | 血清非靶向 LC-MS + GC-MS | MS-DIAL / XCMS R | MZmine 3, asari;MetaboAnalystR, GNPS | 候选 | ⚠ |
| 11 | TMT Bulk 蛋白质组 | FragPipe-TMT + MSstatsTMT R | MaxQuant;limma | 候选 | ⚠ |
| 12 | 跨组学纵向整合框架 (已专题深化 ↓07) | MOFAcell Py + DIABLO R | MOFA+/MEFISTO, SNF(snfpy);WNN/totalVI 不适用 | 专题结论 | ✓ |
| 13 | 富集 / 通路 / 基因集 | decoupleR / clusterProfiler / fgsea R | GSEA;AUCell + GSVA(保留) | 保留+补充 | ⚠ |
| 14 | 铁死亡 / 特定机制基因集 | FerrDb V2 + AUCell 打分 | MSigDB HALLMARK_*;WikiPathways | 候选 | ⚠ |
| 15 | 药物重定位 / 逆转打分(CMap) | signatureSearch R / cmapPy Py + LINCS L1000 | CLUE / connectopedia | 候选 | ⚠ |
| 16 | 发表级矢量图表(SVG/PDF 300DPI) | ggplot2+ComplexHeatmap R / matplotlib Py | patchwork, UpSetR;Cytoscape/igraph | 补充自研 SVG | ⚠ |
| 17 | 可重复流程与环境 | Snakemake(保留)/ nf-core | Nextflow;Docker/Singularity/Apptainer + conda 锁定 | 保留合理 | ⚠ |
注:#13 的 AUCell/GSVA、#17 的 Snakemake、#6 的 RCTD 等"保留"结论中,仅 RCTD 与去卷积梯队经本轮验证(✓);其余"保留合理"为基于来源的合理判断但未单列对抗论断(⚠)。
03已验证选型详情(9 条综合论断)
以下每条均附置信度、投票结果与逐字证据引文。原文关键引文以 蓝色斜体标注。
保留 RCTD(最优梯队·最快·CPU 并行),并同时跑 cell2location 交叉比对
两个独立同行评审基准一致将 RCTD 与 cell2location 列为去卷积最优梯队;针对 55μm 高混杂胎盘 spot 可补充 Tangram/CARD,而 SPOTlight 整体较弱。
Harmony 对高复杂度胎盘整合可能次优;考虑升级 scVI/scANVI 或 Scanorama
权威 scIB 基准(16 方法/14 指标)显示 Harmony 在复杂真实数据非顶档;EVT/SCT 高异质亚群建议 scVI/scANVI(有标签时)或 Scanorama(检测稀有亚群)。但 11 例样本可能属"较小任务",升级需结合真实细胞数实测。
注:"有标签时 scANVI/scGen 必胜"这一更强论断被 3 票否决,故应表述为综合候选而非单点断言。
scvi-tools 活跃维护,最新稳定版 v1.4.3(2026-05-12)
承载 scVI/scANVI 的 scvi-tools 版本不过时、可放心采用为整合主用工具。
对比:项目当前 Scanpy 1.9.8 / squidpy 1.2.3 偏旧,建议评估随 scverse 生态升级以与 scvi-tools 1.4.3 配套。
同病种 PE 论文用「相邻切片 + landmark spot-match」,而非 elastix 同切片配准
一篇晚发型 PE 胎盘空间多组学论文并未在同一切片做图像配准,而是用相邻切片(~10μm)+ 自研 landmark 点对点匹配(≥6 点、缩放+旋转坐标变换)。这是与本项目同场景的直接实证,当前 elastix 仿射+B-spline 同切片设定面临反例。
差异说明:该论文用 DESI-MSI(本项目为 MALDI-MSI),但本论断只涉及配准方法、与电离平台无关。
MALDI-MSI 用 Cardinal 3 重建+聚类,pySM/METASPACE 做 FDR 控制的代谢物注释
开源 Cardinal 3(R/Bioconductor)用于离子图像重建/背景扣除/聚类,pySM(EMBL Alexandrov 团队,METASPACE 注释引擎)用于代谢物注释——同病种 PE 胎盘空间代谢组研究已采用,填补项目 MSI 空白。
限定(故中置信):该研究用 DESI 而非 MALDI;SmetDB 为商业内部库(非开源),开源结论仅限 Cardinal/pySM。
无单一最优、依平台而定;Visium 上 GraphST 领先,BANKSY 统一分型+分域
10X Visium 上 GraphST(ARI=0.552) > STAGATE(0.515) > CCST(0.481);BANKSY 以单一算法统一"细胞分型 + 组织域分割",多个基准胜过 Giotto/BayesSpace/SpaGCN/SpiceMix,是替换/增强 SpaGCN 的强力候选。
注:"GraphST DLPFC ARI=0.633、GraphST>BayesSpace>SpaGCN>STAGATE"被 3 票否决——请采用 gkaf303 的 0.552/0.515/0.481。
CellRank 组合速率核与连通核、在高维基因空间计算转移矩阵,适合 EVT vs SCT 分化
CellRank 比"仅把速率投影到低维嵌入"更鲁棒,适合滋养层双主线分化轨迹分析。
流程应纳入双细胞剔除(改善下游 DE/HVG/聚类/轨迹);工具倾向 scDblFinder
双细胞剔除可测量地改善下游分析;具体工具排名随时间更新,应倾向 scDblFinder(AUPRC/AUROC 与速度领先),而非 2021 基准偏好的 DoubletFinder。
时效性:该论文"DoubletFinder 准确率最佳/前二推荐"的具体排名被 3 票否决——后续独立基准(Germain et al., scDblFinder, F1000Research)显示 scDblFinder 平均 AUPRC/AUROC 最高且更快。
Vento-Tormo 2018(Nature)是滋养层/母胎界面注释的直接参考图谱
~7 万细胞早孕母胎界面单细胞图谱(配套母血+蜕膜),含 EVT/SCT,是注释胎盘细胞类型的直接相关参考;亦是 CellPhoneDB 的来源(与阶段 #5 细胞通讯天然契合)。
限定:该图谱为早孕组织,而 PE 常用足月胎盘,跨孕期注释迁移有 caveat。
04当前锁定工具栈:验证结论
对 tools_memorandum.md V1.2 与 算法设计说明书 V1.1 中已锁定工具的逐项裁决。
✅ 保留 KEEP
- RCTD(spacexr 2.2.0,multi 模式)✓验证
- Scanpy 单细胞主流程
- squidpy 空间转录组
- AUCell + GSVA 通路评分
- WGCNA 数据驱动共表达轨道
- Snakemake 流程编排
- CMap / NCS<-0.6 药效逆转原理
🔼 升级 / 调整 UPGRADE
- 批次整合:Harmony → 评估 scVI/scANVI/Scanorama(按细胞复杂度与样本规模)✓验证
- 新增双细胞剔除:scDblFinder ✓验证
- 版本:Scanpy 1.9.8 / squidpy 1.2.3 偏旧 → 随 scverse 升级以配套 scvi-tools 1.4.3
🔁 替换 / 重评估 REASSESS
- 空间配准:elastix 同切片仿射+B-spline → 评估 相邻切片 landmark spot-match(同病种实证)✓验证
- 空间域:SpaGCN → BANKSY / GraphST ✓验证
➕ 填补空白 ADD
- MSI 处理/注释:Cardinal 3 + pySM ✓验证
- 注释参考:Vento-Tormo 2018 图谱 ✓验证
- 轨迹:CellRank ✓验证
- 整合/蛋白/代谢/药物:见「待补空白」⚠候选
05避坑:被对抗式验证否决的论断
以下 6 条候选论断在 3 票核查中被否决(不足 2/3 支持或遭多数反驳),请勿采用。列出以防误用网络上常见但已过时/过强的说法。
❌ "Harmony 适合 11 例中等复杂度,保留即可" 1-2 否决
不能据此简单保留 Harmony;scIB 显示其在复杂任务非顶档,需结合细胞复杂度评估升级。
❌ "Tangram + Cell2location 最稳健最准" 0-3 否决
过强表述。最优梯队应为 RCTD/cell2location(并列 CARD/Tangram),而非单点钦定。
❌ "有标签时 scANVI/scGen 必胜" 0-3 否决
不要单点断言 label-aware 必胜;应表述为 scVI/scANVI/Scanorama 综合候选。
❌ "DoubletFinder 准确率最佳"(两条) 0-3 否决
2021 基准排名已过时。倾向 scDblFinder(AUPRC/AUROC 与速度领先)。
❌ "GraphST DLPFC ARI=0.633;GraphST>BayesSpace>SpaGCN>STAGATE" 0-3 否决
数值与排序错误。采用 NAR gkaf303 的 Visium 数值:GraphST 0.552 > STAGATE 0.515 > CCST 0.481。
06待补空白:候选工具(本轮未对抗验证)
🧩 跨组学纵向整合框架 已深化 ↓07
把 scRNA / 空转 / 蛋白 / 代谢收敛到统一隐因子或机制模块——项目"跨组学机制模块筛选"目标的核心。已于第 07 节用第二轮 deep research 专门深化(MOFAcell 主 + DIABLO 辅 + 小 n 稳健性纪律)。
- MOFA+ / MEFISTO(bioFAM/MOFA2,R/Py)——多组学因子分解;MEFISTO 加空间/时间协变量
- mixOmics-DIABLO(R)——有监督多组学判别
- Seurat WNN / totalVI / multiVI——单细胞多模态
- SNF(相似网络融合)
💊 药物重定位 / 逆转打分 候选
外植体数据未交付时的现代 CMap 实现与公共参考。
- signatureSearch(Bioconductor,R)——GESS + FEA 一体化
- cmapPy(cmap/cmapPy,Py)——GCTx/L1000 读写
- LINCS L1000 / CLUE 公共参考库(无外植体数据时的替代)
🧪 TMT Bulk 蛋白质组 候选
- 搜库/定量:FragPipe-TMT(Nesvilab)/ MaxQuant
- 差异:MSstatsTMT / limma(R)
注:项目实际蛋白组已有 DIA-NN gg_matrix(见交付站),TMT 工具按是否需重搜库取舍。
🩸 血清非靶向 LC-MS + GC-MS 候选
- 峰提取:MS-DIAL / XCMS(R)/ MZmine 3 / asari
- 注释/统计:MetaboAnalystR / GNPS
🔗 细胞间通讯 候选
- CellPhoneDB(与 Vento-Tormo 母胎界面天然契合)
- LIANA(多方法共识)/ CellChat / NicheNet
📊 富集 & 发表级图表 候选
- 富集:decoupleR / clusterProfiler / fgsea(保留 AUCell/GSVA)
- 制图:ComplexHeatmap(jokergoo/ComplexHeatmap)/ ggplot2+patchwork / matplotlib — 原生 SVG+PDF 300DPI,建议替代部分自研客户端 SVG
07⭐ 专题深化:跨组学整合框架(第二轮 deep research)
针对上一轮标记的头号空白——跨组学整合框架,单独再跑一轮更深、更挑剔的调研。105 智能体 / 5 角度 / 23 来源 / 114→25→23 条确认论断 / 3 票对抗式验证。本轮把项目真实约束(样本层 n=8 配对、Python-only、机制模块目标、已验证"分泌/抗血管轴"信号)全程钉入评估。
落地建议(n=8 · Python 优先 · 机制模块)
🥇 数据驱动主框架:MOFAcell Py
multicellular factor analysis:每个 view = 一种细胞类型的样本级 pseudobulk → 分解出跨细胞类型的"多细胞转录程序"潜因子,最契合"机制模块筛选"目标。无监督拟合,疾病标签事后用 Kruskal-Wallis 关联。纯 Python 路径(liana-py),无需 R。与现有 WGCNA 数据驱动轨道并行/增强,而非替换。
repo:eLife 93161 · saezlab/MOFAcellulaR(R) · liana-py mofacellular(Py)
🥈 监督验证轨道:DIABLO R
mixOmics 的有监督 N-integration:多块 sPLS-DA + 设计矩阵,针对 PE/对照分类结局做整合,稀疏特征选择利于可解释机制。监督整合基准(bbae331 2024)中位 MCC 最高(0.52)、被荐为通用兜底。利用已知标签,小 n 下比无监督更可控。代价:需加装 R。
repo:mixomics.org/mixdiablo · 基准 BiB 2024 bbae331
✅ 实测落地验证(已在 server199 真实数据上跑通)
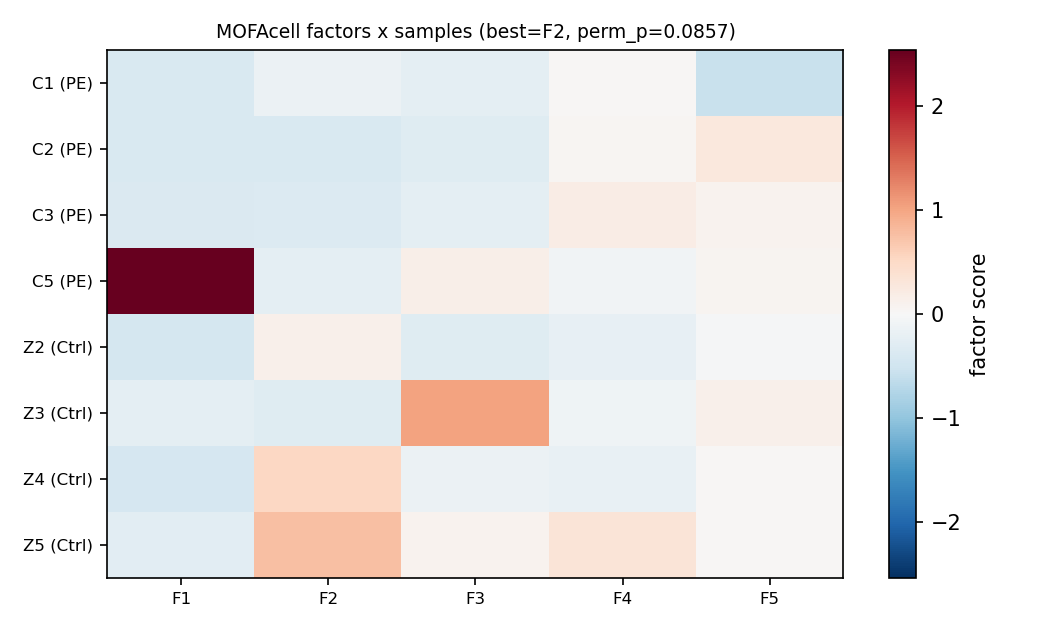
实测结论 SUGGESTIVE
- 最佳分组因子 F2:置换 perm_p=0.086(未过 0.05;n=4v4 理论下限就是 0.029)、LOO 载荷稳定性中位 |r|=0.91、符号一致 8/8 → 因子稳健、但分组显著性仅"suggestive"。
- 跨组学复现「干扰素/炎症轴」:CXCL9/10、IDO1、HLA-G、CD74 在多细胞类型正载荷,蛋白组 CD74 第 97 百分位 → 已 FDR 确认的这条轴获得跨组学加权佐证。
- 「分泌/抗血管轴」较弥散:PAPPA2/LEP/CRH 在基质/免疫区为正,但 SCT 内 HTRA4/LEP 方向不一致,未形成单一干净因子。
- n=8 脆弱性被实证:方差最大的 F1 = 单样本离群(C5=2.54,余者≈−0.3),F3 = Z3 离群——正是研究警告的小样本伪信号。
结论:整合层佐证了干扰素轴的跨组学收敛,但如研究所料,n=8 下它只是假设生成 / 佐证,不凌驾于样本级 DESeq2 + GSEA 结论。
方法类别对比
| 方法类别 | 推荐主用 | 语言 | n=8 可靠性 | 对本项目 |
|---|---|---|---|---|
| 跨细胞类型·多病例程序 | MOFAcell | Py | 需稳健性检验 | 首选·机制模块 |
| 有监督多块判别 | DIABLO (mixOmics) | R | 小n较可控·须CV | 监督验证轨道 |
| 无监督因子分解(样本层) | MOFA+ / MOFA2 | PyR | n<15 作者称"无用" | 慎用·须稳健性 |
| 空间/时间感知因子 | MEFISTO | Py | 仅空间协变量时 | 对静态病例/对照无用 |
| 相似网络融合 / 整合聚类 | SNF (snfpy) / NEMO / iClusterBayes | Py/R | 8 例分层意义有限 | 至多探索性辅助 |
| Python 多模态容器 | muon (承载 MOFA) | Py | — | 可作统一容器 |
| 同细胞多模态 | Seurat WNN / totalVI / multiVI | Py/R | — | ❌ 不适用(需同细胞共测) |
| 小 n 稳健替代(待验证) | AJIVE | Py | 基准称小n最稳/不过拟合 | 建议作交叉验证基准 |
已验证论断详情
MOFAcell 把 MOFA 改造为跨细胞类型样本层整合,分解出"多细胞转录程序"
每个 view = 一种细胞类型按样本聚合的 pseudobulk;无监督分解出共享潜空间中的多细胞程序,疾病标签事后关联——正对应"用 scRNA pseudobulk 跨亚群发现协同机制程序"。与现有 WGCNA 数据驱动轨道并行/增强而非替换。
边界:演示数据为人心梗(~27 样本,分组 13/9/5);小 n 警示仍适用(示例 16–79 样本)。
n=8 远低于 MOFA 可靠区;因子必须配稳健性检验
MOFA 作者称因子分析"至少需 >15 样本";独立基准在 n=53 即见 MOFA/sparse mCCA 过拟合、n≤50 普遍不稳,而 AJIVE 在小 n 最稳。本项目 n=8 明确处于不可靠区。
MOFA 以"样本"为共享维度,TCGA 式队列整合,自动处理缺失模态
数据模型是共享样本/观测索引的矩阵(非同细胞共测),能处理不同特征数、自动忽略缺失值(无隐式插补),匹配本项目样本层场景。
MOFA 全栈有纯 Python 实现,无需安装 R
核心训练在 mofapy2(纯 Python/NumPy,pip install mofapy2);R 包 MOFA2 仅为可选封装。scverse 的 muon(100% Python,NumFOCUS)内置 mu.tl.mofa();MOFAcell 经 liana-py 提供 Python 路径——满足 pe_env 仅 Python 的约束。
DIABLO 是有监督多块判别,针对 PE/对照结局做样本层整合,基准 MCC 领先
mixOmics 的 N-integration(同一批样本上不同检测),多块 sPLS-DA + 设计矩阵最大化跨组学协方差,稀疏特征选择利于可解释机制。监督基准中位 MCC 最高、被荐为通用兜底。宿主语言为 R。
边界:基准在 TCGA 规模下做 5 折 CV,未验证 n=8/组可靠性。
MEFISTO 仅在样本有连续空间/时间协变量时增值,对静态病例/对照无用
MEFISTO 是 MOFA 框架内加高斯过程建模空间/时间依赖的扩展(同为 Python)。但需连续协变量驱动 GP 先验;分类的病例/对照标签不能驱动 → 退化为普通 MOFA。仅可能用于 MALDI-MSI/Visium 的样本内空间整合,且本项目缺切片图像使应用受限。
整合更多组学并不一定更好,反而可能损害性能
两篇独立同行评审基准(任务不同)一致:加入更多数据类型常使整合性能下降(噪声相消、冗余、统计挑战)。本项目应谨慎、有选择纳入模态,优先信号已验证、样本重叠好的组学。
癌症亚型基准:NEMO/SNF 居前,但排名场景依赖;n=8 分层意义有限
十方法基准中 NEMO 总体最佳、其次 SNF/iClusterBayes/LRAcluster;但排名随场景而变、存在跨设计乐观偏差。SNF 有 Python 实现(snfpy)适配 pe_env,但 8 例做无监督分层统计可行性弱,至多作探索性辅助。
同细胞多模态法不适用;小 n 下多数无监督整合需高度保留
- ❌ Seurat WNN / totalVI / multiVI——需同一细胞共测多模态,本项目是跨独立检测的样本层整合,不适用。
- ⚠ MOFA/MOFA+ 无监督因子——n=8 远低于可靠区,无稳健性检验则不可信。
- ⚠ SNF/iClusterBayes 整合聚类——8 例分层统计意义有限。
- ⚠ MEFISTO——对静态病例/对照设计退化为普通 MOFA,无增值。
- ✅ 澄清:"muon 仅面向同细胞数据"的说法本轮被否决(1-2)——muon 可作 Python 统一容器承载样本层 MOFA。
08这些调研到底有没有价值?(做 vs 不做)
诚实评估:相对于"不做调研、直接照已锁定方案开干",这两轮 deep research + 一次实跑,对本项目改变了什么。结论先行——净值主要在 避坑省时、防小样本过度解读、决策有据、真实信号加固 四类;它没有产生新生物学结论,核心结论仍来自样本级 DESeq2 + GSEA。
| 环节 | 不做调研的默认路径 | 调研改变了什么 | 价值 |
|---|---|---|---|
| 空间配准 | 按算法书用 elastix 同切片仿射+B-spline | 同病种 PE 胎盘论文证明应是相邻切片 + landmark spot-match;当前范式有同场景反例 → 避免在错误方案上耗时(且呼应项目缺切片图像的现实) | 避坑 |
| 跨组学整合 | 易顺手抓 Seurat WNN/totalVI(最流行的"多模态"法) | 厘清本项目是样本层 n=8、非同细胞共测 → 这类法结构上不适用,避免一条走不通的路;正解是 MOFAcell/DIABLO | 避坑 |
| 小样本解读 | 在 n=8 上跑整合、得到因子就当真写进交付 | 给出 MOFA 自述 ">15"、独立基准 n≤50 过拟合的硬证据,并实跑证明 F1/F3 是单样本离群因子 → 避免把伪信号当结论 | 避坑 |
| 双细胞剔除 | 用 2021 流行的 DoubletFinder | 后续基准显示已被 scDblFinder 超越(AUPRC/速度)→ 直接用更优工具 | 省时 |
| 去卷积 RCTD | 保留,但心里没底、可能反复评估 | 两个独立同行评审基准确认 RCTD 属最优梯队 → 保留有据,省去纠结(并加 cell2location 交叉) | 有据 |
| MSI / 代谢注释 | 空白,从零摸索 MSI 处理与代谢物注释 | 同病种 PE 研究已用 Cardinal 3 + pySM → 直接复用同病种范式 | 省时 |
| 细胞注释参考 | 用通用图谱 | 指向 Vento-Tormo 2018 母胎界面图谱(含 EVT/SCT)→ 更贴合的滋养层注释 | 有据 |
| 实跑 MOFAcell | 无跨组学层面的独立佐证 | 实跑显示干扰素/炎症轴跨 6 细胞类型 + 蛋白组收敛(CD74 第 97 百分位)→ 为一条已确认轴加上跨组学独立佐证 | 加固 |
- 没有新生物学:核心结论仍来自样本级 pseudobulk DESeq2 + GSEA;调研的作用是"选对工具、避开坑、加固佐证",而非"发现新机制"。
- 部分结论资深生信本就知道(如 WNN 需同细胞数据);价值在于把这些以证据固化,并捞出没那么显然的(同病种配准反例、DoubletFinder→scDblFinder 的迭代)。
- 空白工具仍是候选:TMT 蛋白搜库、血清 LC/GC-MS、药物逆转等给的是有依据的起点,尚未对抗验证,不是定论。
- n=8 是硬约束,任何工具都改不了——调研在这里的价值恰恰是诚实地划出这条线,而非假装能跨过。
09注意事项与边界
- 时效性:"工具排名"比"是否纳入某步骤"更易过期——双细胞最优工具已从 DoubletFinder 迁移到 scDblFinder;scIB 基准虽仍是领域标准,但 2024–2026 有后续工作精炼其指标。
- 样本量边界:scVI/Scanorama 推荐基于"足够大数据集",本项目约 11 例可能落在"较小任务"区间(scIB 称此时 Harmony 可能有用);升级建议依赖"EVT/SCT 高复杂度"框架,需在真实细胞数上判断,scVI 在 cells<genes 时可能过拟合。
- 平台不匹配:阶段 8/9 最关键的同病种实证用 DESI-MSI(非项目 MALDI-MSI),其 spot-match 针对相邻切片(非同切片);Cardinal/pySM 虽原生支持 MALDI/imzML,但"同病种已验证"强度限于 DESI;该论文 SmetDB 注释库为专有非开源(故 #9 证据等级为 medium)。
- 去卷积平台细节:Stereoscope 在 10x Visium 上具体表现并不差,"优于 Stereoscope"仅在"整体排名"意义上成立。
- 未验证覆盖:第 06 节"待补空白"的工具未获本轮对抗论断覆盖,仅为候选;报告未对其给出实证结论。
- 流程性质:本报告为对 19 条已通过 3 票验证论断的综合,未在综合阶段重新独立检索;置信度基于来源质量评估。
10来源(第一轮 25 篇·精读取证)
项目 2605136 · 子痫前期胎盘多组学整合 — 生信工具选型调研报告。由 Claude Code deep-research 多智能体流程生成:第一轮全栈(107 智能体 / 25 来源 / 19 条确认)+ 跨组学整合专题第二轮(105 智能体 / 23 来源 / 23 条确认),均经 3 票对抗式验证,2026-06-17。
本报告为工具选型决策支持,不构成对任一工具的最终背书;落地前请结合项目真实数据与专家评审。所有"候选·未验证"项需后续专门调研。相关:项目交付站 pe.sinogenomics.com。